AI|ffinity Lead Design
Our deep-learning algorithm, deepScaffOpt, stands as a high-accuracy solution for binding affinity prediction and potential inhibitor ranking, distinguished as the most precise ligand-based algorithm in the recent D3R Grand Challenge
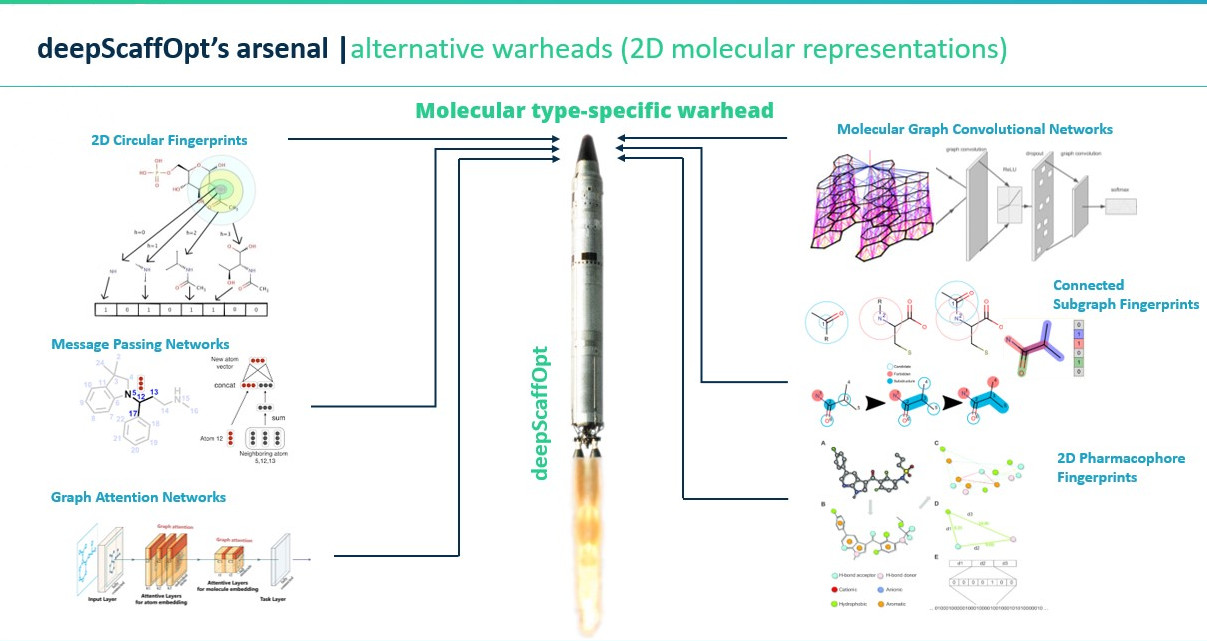
deepScaffOpt's structure comprises two main components: the "main engine" and the "warhead". The warhead is molecular-type specific, with versions designed for various structures like macrocycles and covalent inhibitors. This component transforms the 2D chemical structure of the training molecules into vector form. The main engine, consisting of numerous dense network layers and a loss function specialized for affinity ranking, then processes these vectors. Multiple warheads can be attached to the same main engine for enhanced performance, resulting in a target-specific model capable of rapidly predicting affinities for hundreds of molecules.
Discover what AI|ffinity can do for your company
deepScaffOpt’s Special Features:
- Independence from Receptor 3D Structure: Beneficial but not necessary.
- Diverse Molecular Representations: Over 60 types catering to varied drug design requirements.
- Exclusive 1D NMR Enhancement: Enhances 2D or 3D molecular representations using NMR screening data, especially if receptor structure is identified. An industry-exclusive feature.
- Tailored Deep Learning Approach: Includes custom neural network layers, a unique loss function, and a distinct early stopping method for precise binding affinity predictions.
- Streamlined Installation: Packaged within a docker image containing all essential dependencies, ensuring a hassle-free one-click setup.